VASP 输入文件
VASP 输入文件
介绍
- 包括 INCAR、POSCAR、KPOINTS 和 POTCAR 4 个输入文件
参考资料
内容精简,值得阅读参考:VASP 计算基础与结构优化过程
vasp中影响并行效率的三个变量KPAR,NPAR,NCORE - 第一性原理 (First Principle) - 计算化学公社
是否开启自旋极化
VASP 命令 - 仿真软件说明 - 邱新龙 simulation(VASP 相关 tags 的总结很凝练)
POSCAR
构型文件;生成 POSCAR 文件是 VASP 计算的起点
需至少包含体系的几何信息(晶格常数、基矢、元素种类及对应原子数目)和原子位置(以及 AIMD 计算时原子的初始速度(不常用));可手动生成,也可从一些在线晶体学数据库(Material Project、aflow、icsd 等)中获取
- 第 1 行:Comment line 注释行;可对体系进行描述,也可空着
- 第 2-5 行:Scaling factor and lattice,缩放因子和基矢;与体系的晶格常数符合即可;第二行值如果为负数,表示体积
- 第 6-7 行:Ion species and numbers,元素种类(VASP4 可没有该行)及对应原子数目;元素种类的顺序需与 POTCAR 文件中的一致;
- 第 8-N 行:Ion positions 原子坐标;Direct/D 表示分数坐标,Cartesian/C 表示笛卡尔坐标(若第 8 行是 Selective Dynamics,可简写成 S/SD;原子位置后面每个方向需添加 T/F,表示是否对 x y z 方向进行固定;默认值为 T,表示该方向可运动,F 表示固定;CONTCAR 中也对应会有该信息)
- 原子坐标信息之后是原子的初始速度信息(一般可不用设置)
注意事项:
- VASP 根据 POSCAR 文件确定体系的对称性。原子位置精度不够(位数太少)是一个常见错误。为更好地利用 VASP 中的对称性,强烈建议在 POSCAR 文件中指定至少 7 位有效数字的原子位置(和晶格参数,最好多一些)
1 | # 示例 |
POTCAR
VASP6 与 VASP5 相比,赝势基本不变,只有小部分变动(如 O_h,5.4 为 700.0 eV,6.4 为 765.519 eV 等:求助!跪求一套VASP 6 版本的赝势 - 第一性原理 (First Principle) - 计算化学公社
Available pseudopotentials - VASP Wiki:含 PBE52、PBE54、PBE64 赝势介绍,赝势加后缀之间的区别
赝势文件;包含计算体系中每种元素的赝势(元素种类的数量大于 1,只需将各元素种类的 POTCAR 文件依次连接起来即可,与 POSCAR 文件中元素种类顺序对应)
赝势目录一般含 LDA,PBE,和 PW91 三个子目录
PBE 赝势可分为:无后缀、_pv、_sv、_d 和数字后缀,即 semi-core 的 p、s、d 层电子当做价电子处理
1 | _sv # p 和 s 层电子考虑为价电子;ENMAX 比 _pv 高 |
注意事项:
- 赝势种类:
- 模守恒赝势:Norm conserving PP;所需截断能高,计算精度高
- 超软赝势:Ultrasoft PP;所需截断能较小,计算效率高
- 缀加平面波赝势:Projector Augmented Wave PP;常用
- 赝势目录中每个类型的泛函目录中有一个 data_base 文件,里面包含每个赝势对应元素 3 种可能结构的基态能量数据
- PSCTR 文件:控制赝势生成文件:PSCTR
- VASP中的赝势 - 计算材料学
- VASP5.4 版本,W_sv 替代 W_pv
- 赝势种类:
pymatgen 与 VASP 推荐赝势之间的差异(部分)
| 元素 | MPRelaxSet 推荐 | ENMAX | 价电子 | VASP 推荐 | ENMAX | 价电子 |
|---|---|---|---|---|---|---|
| B | B | 319 | 3 | B | 319 | 3 |
| C | C | 400 | 4 | C | 400 | 4 |
| Al | Al | 240 | 3 | Al | 240 | 3 |
| Si | Si | 245 | 4 | Si | 245 | 4 |
| Sc | Sc_sv | 223 | 11 | Sc_sv | 223 | 11 |
| Ti | Ti_pv | 222 | 10 | Ti_sv | 275 | 12 |
| V | V_pv | 264 | 11 | V_sv | 264 | 13 |
| Cr | Cr_pv | 266 | 12 | Cr_pv | 266 | 12 |
| Fe | Fe_pv | 293 | 14 | Fe | 268 | 8 |
| Co | Co | 268 | 9 | Co | 268 | 9 |
| Ni | Ni_pv | 368 | 16 | Ni | 270 | 10 |
| Zr | Zr_sv | 230 | 12 | Zr_sv | 230 | 12 |
| Nb | Nb_pv | 209 | 11 | Nb_sv | 293 | 13 |
| Mo | Mo_pv | 225 | 12 | Mo_sv | 243 | 14 |
| Hf | Hf_pv | 220 | 10 | Hf_pv | 220 | 10 |
| W | W_sv | 223 | 14 | W_sv | 223 | 14 |
| Y | Y_sv | 203 | 11 | Y_sv | 203 | 11 |
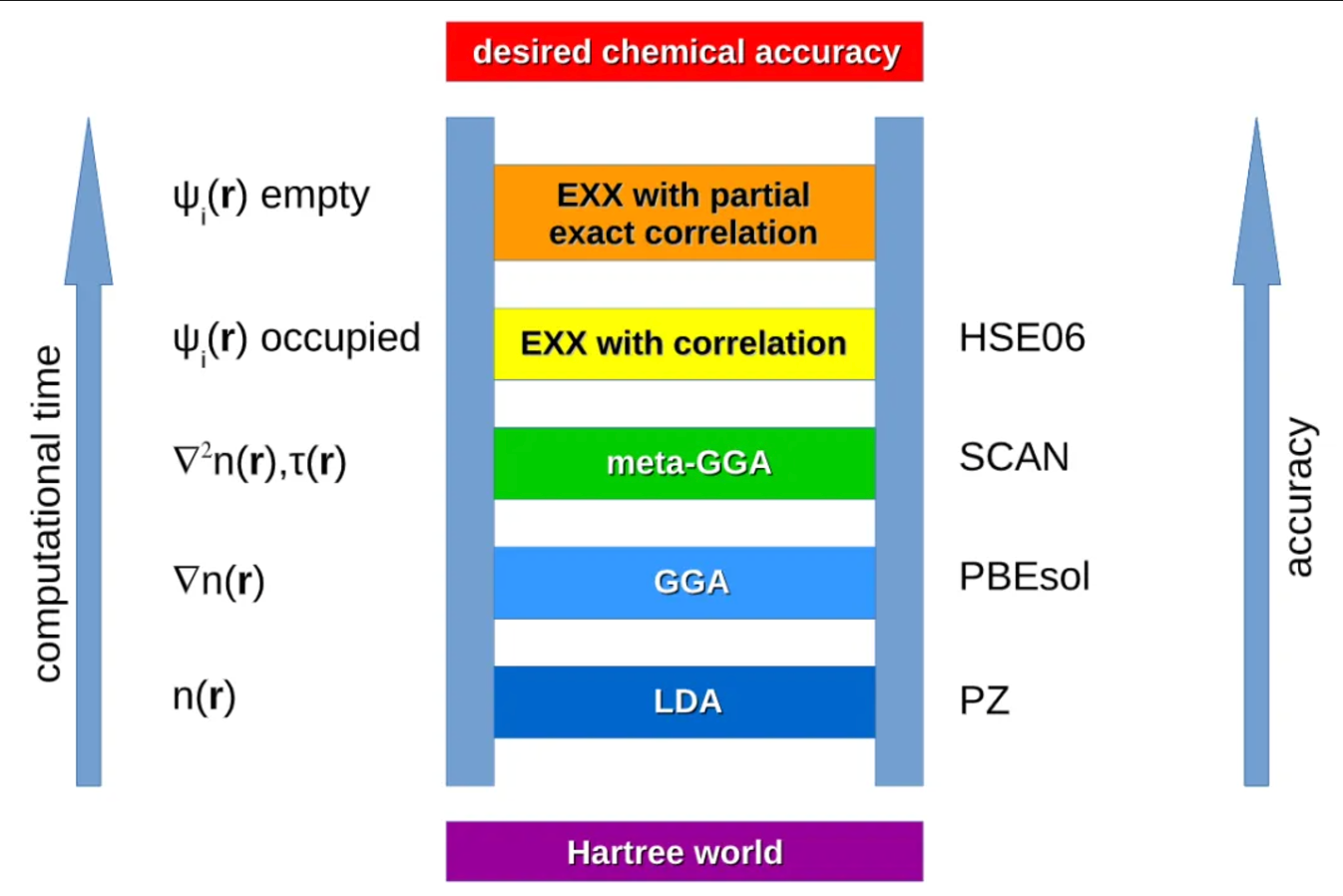
- Jacob 天梯示意图
- POTCAR 文件内容示例:
1 | PAW_PBE Cu 22Jun2005 |
KPOINTS
论文 - Rapid generation of optimal generalized Monkhorst-Pack grids - CMS
INCAR 文件中的 KSPACING 和 KPOINTS 文件同时存在时,KSPACING 设置不生效
设置布里渊区 K 点网格采样大小或计算能带结构时沿高对称方向的 K 点
对 K 点进行收敛性测试是许多电子最小化计算的基本任务之一
常规 K 点网格
- 第 1 行:注释行,可随便写
- 第 2 行:设置 K 点数目,0 表示 K 点网格自动生成
- 第 3 行:K 点网格划分方式(Monkhorst-Pack 和 Gamma-centerd MP 方法);只认第一个字母,大小写均可
- 第 4 行:3 个方向上具体的网格划分数目
- 第 5 行:shift;一般保持 0 0 0 不变
1 | Regular k-point mesh |
Gamma-centered(以 Gamma 点为中心生成网格) 只是 MP 方法的一种特殊情况
不同晶系下的 K 点生成方式选择:Symmetry reduction of the mesh - KPOINTS - VASP Wiki
- Monkhorst-Pack 网格的收敛速度可能快于 Γ- 中心网格;同时需注意避免使用 Monkhorst-Pack 网格破坏对称性
- 对于 HCP、面心立方结构,K 点生成方式采用 Gamma-centerd(M 平移之后,网格的对称性和晶胞的对称性会出现不匹配的情况,从而导致计算出错)
- 建议一直用 Gamma-centered
- 非六方晶系的计算,若已设置 M 计算了,继续沿用计算就行,没必要改成 G 再重新算一遍
- 若晶胞在某个方向增加了 2 倍,对应方向的 K 点需除以 2(在计算过程中,保持 k*a 保持不变):原因:倒易晶格矢量和实际的晶格矢量之间存在着倒数的关系;选取的晶格越大,倒易晶格矢量越小。用同等数目的 K 点分布到倒易晶格中,网格的密度也会越大,从而造成计算量的增加
自动生成;推荐使用 INCAR 中的 KSPACING 参数
能带计算:当性质依赖于 K 矢量时,通常沿高对称性路径将性质可视化;线模式(line mode)表示在布里渊区用户定义的点之间生成 K 点,最常用的情况是分析能带结构
- 第 1 行:注释行
- 第 2 行:设置 K 点数目,非 0 数字表示每条线之间划分的 K 点数目
- 第 3 行:生成 K 点方式
1 | k points along high symmetry lines |
- K 点密度选择:三个方向的 K 点密度应尽量一致
- 对于原子或者分子的计算,取一个 Gamma 点就够了(1 1 1)
- 表面 Slab 体系,真空层方向的 K 点取 1 即可
1 | # a 为 晶格常数 |
INCAR
核心输入文件:用于指定 VASP 计算的参数、算法和设置
INCAR 准备的原则:越简单越好,不知道的,不理解的就不往里面放
注释
#,可在没有歧义的情况下以不添加#进行注释INCAR 参数类型
- 通用参数:SYSTEM、PREC、ISTART、ICHARG、LWAVE、LCHARG
- 态密度积分相关参数:ISMEAR、SIGMA
- 电子优化相关参数:ALGO、ENLM、NELMIN、ENCUT、EDIFF
- 离子优化相关参数:IBRION、POTIM、NSW、EDIFFG
- 态密度相关参数:LORBIT、EMIN 、EMAX、NEDOS
- 能带相关参数:NBANDS
- 高压相关参数:PSTRESS
对于大体系,建议增加 NELM 和 NSW 数值
注意事项:
- 等号(=)前后可以有空格,也可以没有
- 不要使用 Tab,用空格替换 Tab
- INCAR 中的参数中的 L 开头表示逻辑数
- INCAR 参数名称写错,VASP 会忽略,不影响
- 参数设置的第一个数值为默认值,忽略后续同参数的数值设置
- .FALSE. 可简写为 F,.TRUE. 可简写为 T
SYSTEM
title string,对体系及要执行的计算进行注释说明
默认值:unknown system
可随便写;该参数可有可没有
ISTART
初始化轨道;确定是否读取 WAVECAR 文件
默认值:1(若 WAVECAR 文件存在)否则 0
若无 WAVECAR,设置 ISTART=1/2,不会报错,而是继续算
ICHARG
决定 VASP 如何构造初始电荷密度;其积分值为电子数
默认值:ISTART=0,则 ICHARG=2;否则 ICHARG=0
1 | 0 # 从初始波函数计算电荷密度 |
ISPIN
是否考虑自旋极化(1 不考虑;2 考虑)
默认值:1
若确定研究体系不含磁性,尽量不开启自旋极化(计算量至少是原本的 2 倍以上;但并不会对计算的性质结果产生影响,可检查添加自旋后的计算磁矩是否接近 0);若含磁性,可先不开启自旋极化,进行结构优化,将其生成的电荷密度文件作为后续开启自旋极化计算的电荷密度输入
需考虑自旋极化的几种情况:
- 含 Fe,Co, Ni 的体系
- 体系具有磁性:顺磁,铁磁,反铁磁等
- 关注体系的电子性质且不确定时,建议加上
LCHARG
决定电荷密度是否写入 CHGCAR 和 CHG 文件中
默认值:.TRUE.
LWAVE
决定运行结束后波函数是否写入 WAVECAR 文件中
默认值:.TRUE.
LREAL
决定投影算子是在实空间还是在倒空间求值
默认值:.FALSE.
体系超过 30 个原子,推荐使用实空间投影 scheme,且使用 LREAL=Auto
LREAL=Auto 总是会导致小误差(不一定可忽略;通常是每个原子的恒定能量偏移);关注能量差,计算时使用相同的设置(ENCUT、PREC、LREAL);弛豫结构,最后静态计算时,使用 LREAL=.FALSE. 获取精确能量;PREC=Accurate,误差通常小于 1meV/atom
1 | .FALSE. # 倒空间 |
PREC
计算精度;设置截断能、FFT grids 和 the projectors in real space ROPT 的精度的默认值
默认值:Normal;推荐使用 Normal 或 Accurate
1 | Normal # 适用于大多数常规计算 |
ENCUT
平面波(基矢集)截断能;收敛性测试指标之一
默认值: POTCAR 文件中最大的 ENMAX 值
原则上可不进行具体设置,会默认读取 POTCAR 中的 ENMAX 数值(建议具体设置)
ENCUT 值确定好后,在计算中需保持不变
ALGO
决定电子最小化算法,或选择 GW 计算类型
默认值:Normal
blocked-Davidson (DAV) 算法对应于 IALGO=38;RMM-DIIS (RMM) 算法对应于 IALGO=48
建议通过 ALGO 选择算法而非 IALGO
1 | Normal # blocked-Davidson 算法 |
ISMEAR
费米狄拉克分布示意图及展宽处理
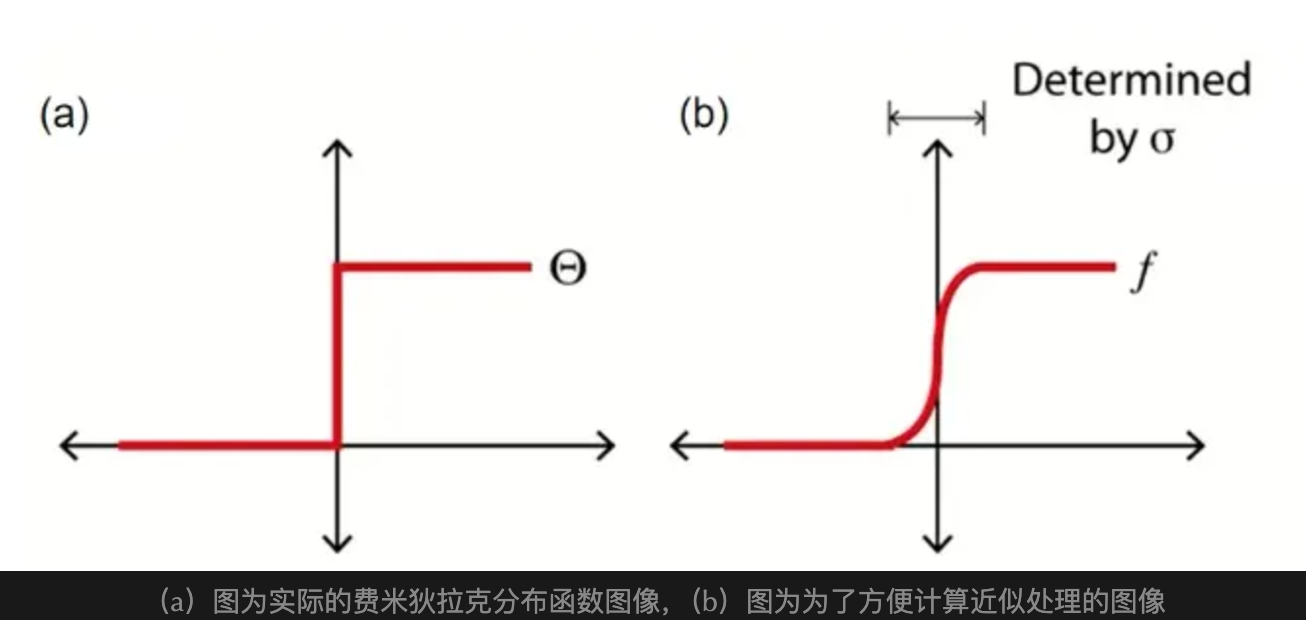
轨道分数占据(电子态占据数,值在 0-1 之间)的展宽(平滑处理)方法
默认值:1
ISMEAR=0 在大多数情况下会得到非常合理的结果
原子受力和应力与
free energy一致,而不是sigma->0外推能量,因此需确保原子受力和应力收敛于对应的 SIGMA 值ISMEAR=-5 时,SIGMA=0,费米狄拉克分布,不会有展宽;电子态的占据是整数占据(0 或 1);无平滑处理,DOS 绘制普遍毛刺较多
对体系没有先验认识,总是使用 ISMEAR=0 + SIGMA=0.03-0.05
DOS 和非常精确的总能计算,使用 ISEMAR=-5
- 缺点:对于金属的力和应力张量,误差可能高达 5-10%
对于半导体或绝缘体,使用 ISEMAR=-5,若胞太大或只使用 1-2 个 K 点,使用 ISMEAR=0 + SIGMA=0.03-0.05
- 避免使用 ISMEAR>0,因为经常会导致错误的结果(某些态的占据可能小于 0 或大于 1)
对于金属中的力、声子频率计算,使用 ISMEAR=1 或 ISMEAR=2,SIGMA 合理值通常为 0.2(默认值)
- 引入 SIGMA 展宽后,使得 OUTCAR 中的 T*S 熵项不为 0(该熵值不是由一定的温度带来的,而是数学处理的结果)
- 推荐使用 ISMEAR>0(总能也能精确描述),需仔细选择 SIGMA 的值。值太大可能导致不正确的总能,太小需要更密的 K 点;应尽可能大,使 OUTCAR 文件中的
entropy T*S项可忽略(小于 1meV/atom)
Methfessel-Paxton order N 示意图:Methfessel-Paxton Approximation to Step Function
1 | N # N 为数字;Methfessel-Paxton order N(默认 1) |
SIGMA
展宽宽度(单位:eV)
默认值:0.2
对于金属,默认 0.2,但通常 0.05 就能满足要求
EDIFF
电子步(the electronic SC-loop)收敛条件
默认值:$10^{-4}$(单位为 eV,而非 eV/atom)
在大多数情况下,收敛速度是二次的,因此额外迭代的成本通常很小;收敛良好的计算,强烈推荐设置 EDIFF= $10^{-6}$;有限差分计算(如声子),为获取精确结果,可能需要 EDIFF= $10^{-7}$;大体系或使用 meta-GGA 泛函,EDIFF= $10^{-8}$ 或 EDIFF= $10^{-7}$ 的收敛条件可能很困难;总体 EDIFF= $10^{-6}$ 可能为最佳设置
NELM
最大电子步步数
默认值:60
通常无需修改默认值;若电子自洽在 40 个电子步内收敛,可能根本无法收敛,此情况应考虑 ALGO 等参数
梅师兄设置:300
NELMIN
最小电子步步数
默认值:2;推荐值设置在 4-8 之间
在 AIMD 中,每个离子步中的电子步可能会很少(1-2 步),需对其进行最小步数限制(可能计算结果更准确)
NSW
最大离子步步数
默认值:0
IBRION=0 时,必须提供 NSW 数值(AIMD 步数)
每个离子步会计算 Hellmann-Feynman 力和应力
IBRION
决定在计算中晶体结构如何变化
默认值:-1(NSW=-1 或 0);0(其他情况,执行 AIMD)
IBRION=-1 时,避免设置 NSW>0, 否则重新计算相同结构 NSW 次
除 0 外,其他算法都最终弛豫到能量局域最小值
结构优化
- IBRION=1,通过之前位置的线性组合来减少力;接近基态的大体系(> 20 个自由度)选择
- IBRION=2,沿搜索方向寻找最佳步长;稳健的默认选择,相比 RMM-DIIS 可能需要更多的迭代步数
- IBRION=3,runs a MD simulation with decreasing velocity of the ions;远离基态的大体系选择,相比其他算法,获取更好的起始点(效率不高;用的情况很少)
阻尼分子动力学(Damped Molecular Dynamics,Damped MD)优化算法是一种结合 MD 和优化技术的方法,用于寻找系统的能量最小状态。这种方法在传统 MD 中引入阻尼项,以加速系统朝向能量更低的状态演化(ChatGPT4 生成)
有限差分(finite differences);密度泛函扰动理论(density functional perturbation theory, DFPT)
1 | -1 # 不更新;离子不移动 |
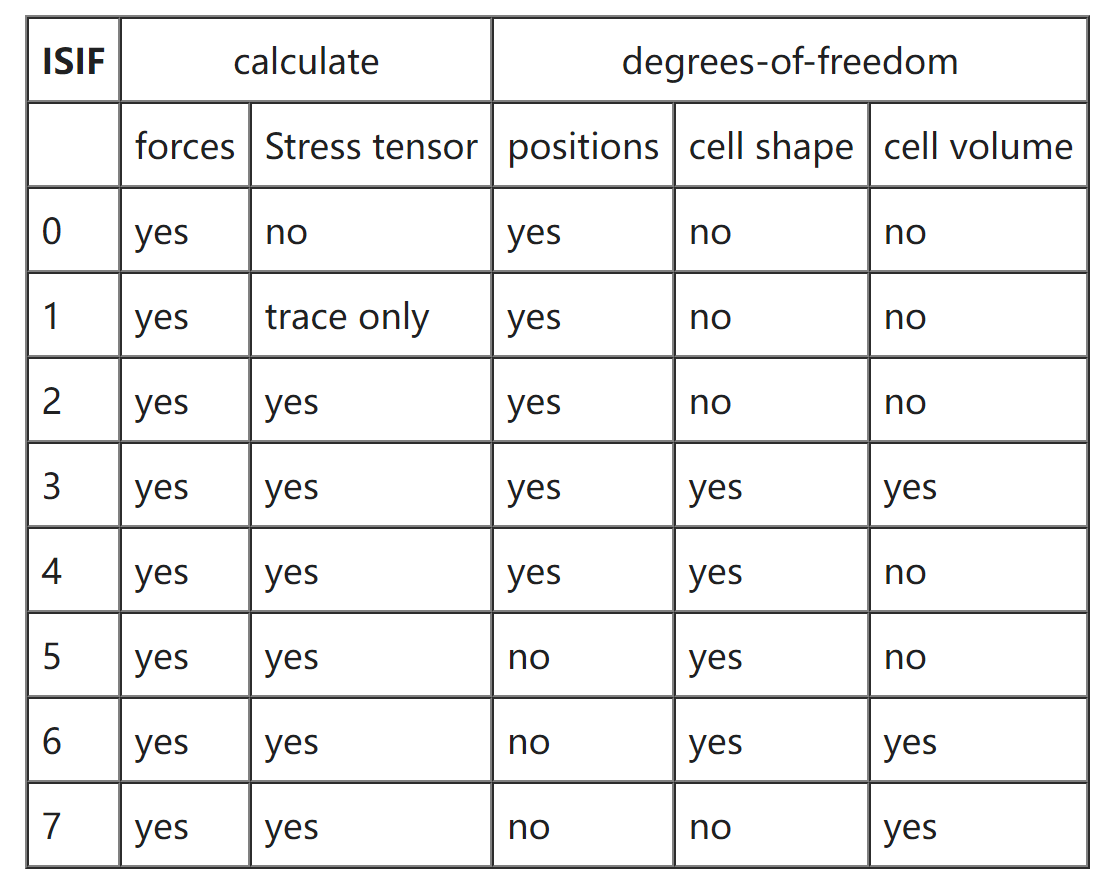
ISIF
决定在弛豫及分子动力学运行中胞的体积、形状或原子位置是否发生改变以及应力张量是否计算
默认值:0(IBRION=0 时);2(其他情况)
常用值:3、2、4
应力张量计算相对耗时,因此在 AIMD 中将其关掉;力总是会进行计算
1 | 1 # 只弛豫原子位置,晶胞体积、晶胞形状不变(不计算应力张量) |
EDIFFG
离子步(the ionic relaxation loop)收敛条件
默认值:EDIFF * 10
EDIFFG 为正值,表示两离子步间的总能变化小于 EDIFFG 时,弛豫结束;为负值,表示受力小于 |EDIFFG|时,弛豫结束(绝对值,更方便的设置);EIDIFF=0 时,运行 NSW 步后,离子步结束
EDIFFG 不用于 AIMD
POTIM
离子弛豫步宽或 AIMD 时间步长
默认值:none(IBRION=0 时必须设置);0.5(IBRION=1, 2, 3,离子弛豫);0.015(IBRION=5, 6)
IBRION=1, 2, 3 时,POTIM 相当于步长的缩放常数;quasi-Newton 算法对该参数值敏感(IBRION=1)
IBRION=5, 6 时,POTIM 相当于每个离子的位移宽度(计算 Hessian 矩阵)
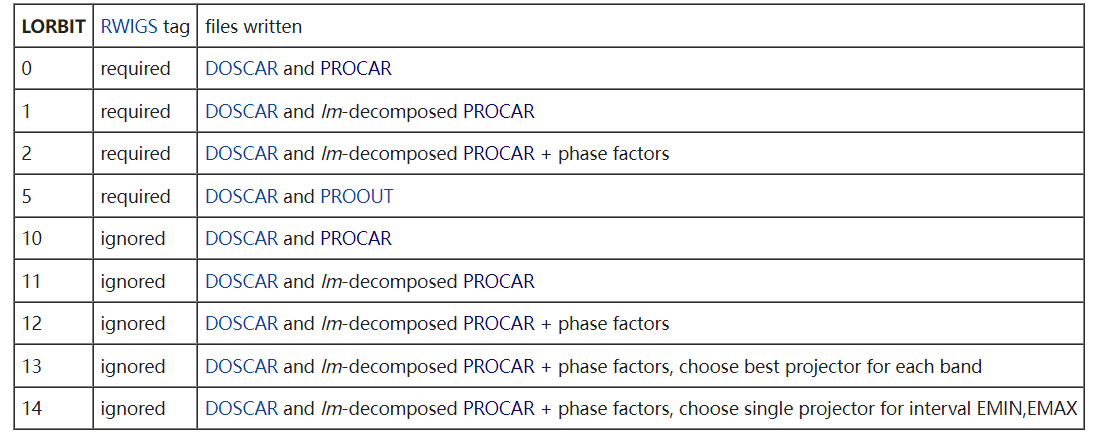
LORBIT
和适当的 RWIGS 一起,决定 PROCAR 或 PROOUT 文件是否被写入。LORBIT>=10 时,不需要 RWIGS 标签
默认值:0
ADDGRID
添加网格
默认值:.FALSE.
若 ADDGRID=.TRUE.,grid 将是 “fine” grid 的 8 倍;有助于降低力噪声;不应在所有计算中将其作为默认的 tag
SYMPREC
决定 POSCAR 文件中的位置精度
默认值:$10^{-5}$
ISYM
决定 VASP 处理对称性的方式
默认值:1(赝势使用 USPPs);3(LHFCALC=.TRUE.);2(其他情况)
1/2/3:对称性打开;-1/0:对称性关闭
与 ISYM=1 相比,ISYM=2 采用了更高效、更节省内存的电荷密度对称化方法,尤其降低了并行版本的内存需求
对于 ISYM=3,VASP 并不直接对称电荷密度。相反,电荷密度是通过对布里渊区不可约部分 k 点处的轨道进行相关对称运算来构建的
ISYM=0,VASP 不使用对称性,但会假定 Ψk=Ψ*-k 并相应减少布里渊区的采样(对应 AIMD 设置,即 IBRION=0)
ISYM=-1,对称性被完全关闭
若对称性打开,则 NWRITE=3 将对称操作写入 OUTCAR 文件
该 tag 的 VASP 官网中还讲解了对称性的相关内容
缺陷(空位、掺杂、表面)计算,设置 ISYM=0 会对最终的计算能量、构型是否有影响(最后的能量和构型均一样,无影响,但更耗时,耗时是原来的 4 倍(粗略测试))
1 | # 测试路径 |
MAGMOM
设置每个原子的初始磁矩
默认值:NION * 1.0 (ISPIN=2)
KSPACING
若 KPOINTS 文件不存在,决定 K 点数目
默认值:0.5(推荐值 0.1、0.15、0.2)
k 点之间允许的最小间距(单位 Å);间距减小,k 点数目增加;与 KPOINTS 文件中的 automatic k-point generation 不完全相同;推荐在 INCAR 文件中使用 KSPACING,避免在 KPOINTS 文件中使用 automatic mode
KGAMMA
决定 K 点是否含 $\Gamma$ 点
默认值:.TRUE.
若 KPOINTS 文件存在,VASP 忽略 KSPACING 和 KGAMMA 参数
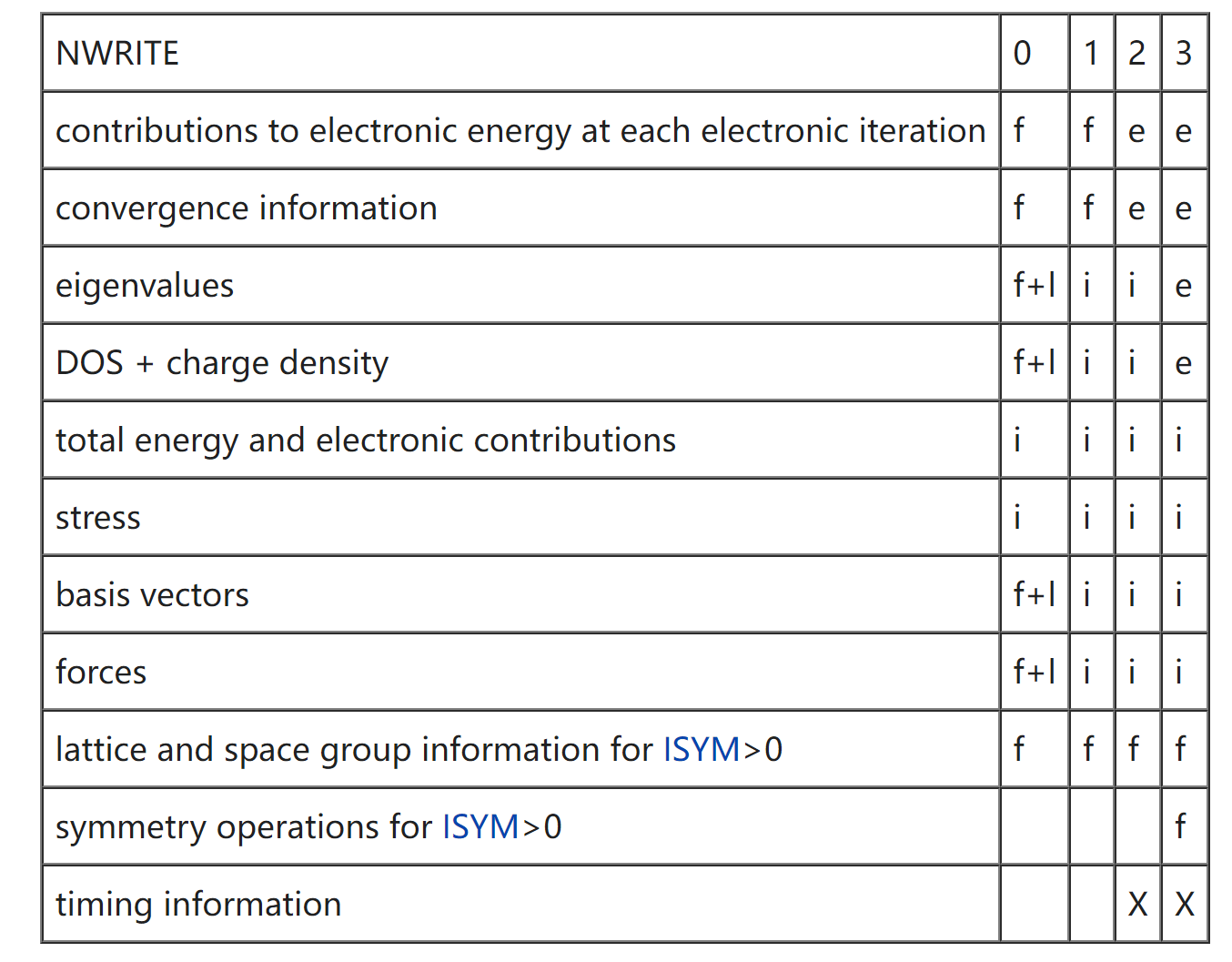
NWRITE
决定往 OUTCAR 文件中写入多少内容
默认值:2;可选值:0-4
f+l 表示第一步和最后一步离子步, f 表示第一个离子步,i 表示每个离子步,e 表示每个电子步,X 表示适用时(when applicable)
长时间的 MD 运行,建议 NWRITE 设置为 0 或 1;短时间运行设置为 2
1 | 0 # 只写入第一步和最后一步的基矢、受力信息(vasprun.xml 中仍含每个离子步的相关信息) |
EMIN & EMAX
- DOS 能量的上下范围
NEDOS
DOS 和介电函数的网格点数目
默认值:301
LAECHG
LAECHG=.TRUE. 时,all-electron 电荷密度将被明确重建并写入文件
默认值:.FALSE.
LAECHG=.TRUE. 时,VASP 会重建三个不同的 all-electron 电荷密度,分别写入 AECCAR0,AECCAR1 和 AECCAR2 文件
- the core density
- the proto-atomic valence density (overlapping atomic charge densities)
- the self-consistent valence density
在 PAW 方法中,”all-electron” 密度不是指所有电子(all electrons)的密度
NFREE
WIP…
LASPH
考虑 PAW spherical 密度梯度相关的 non-spherical 贡献(LDA 和 Hartree potential 通常考虑)
默认值:.FALSE.
利于精确总能计算、能带结构计算(f、所有 3d、第二周期的磁性原子)
对 DFT+U, HSE, metaGGA, vdw-DFT 重要
开启后,会增加计算耗时(增加不算太多?)
LMAXMIX
DFT+U 时,含 d、f 电子的元素值需增大
对非自洽计算能带非常重要
默认值:2
值增大,会增加计算耗时(增加不算太多?)
LVTOT
决定总的局域势是否写入 LOCPOT 文件
默认值:.FALSE.
总的局域势包括:离子、Hatree 和交换关联势
LVHAR
决定局域势是否写入 LOCPOT 文件
默认值:.FALSE.
局域势包括:离子、Hatree 势,不包括交换关联势
用于静电势能的计算
LELF
决定是否生成 ELFCAR 文件
ELFCAR 文件含电子局域化函数(electron localization function, ELF)
若设置了 LELF,则必须在 INCAR 文件中明确设置 NPAR=1
PSTRESS
设置外部压力(Pully Stress,单位 kB)或对应力张量进行修正
默认值:0
NBLOCK
每 NBLOCK 个离子步后,对关联函数和 DOS 被计算,且构型写入到 XDATCAR 文件中
默认值:1
推荐设置成 1, 其计算开销很小;仅在长时 AIMD 或使用 MLFF 时,可将 NBLOCK 提高至 10 甚至 100
使用
ML_MODE = run预测模式时,更倾向于用ML_OUTBLOCKtag
KBLOCK
每 NBLOCK * KBLOCK 个离子步后,对关联函数和 DOS 的平均值写入 PCDAT 和 DOSCAR 文件中
默认值:NSW 参数值
SMASS
控制 AIMD 运行过程中的速度
默认值:-3
对于特定体系,Nosé-mass 的设置应使温度振荡频率与典型的 “声子” 频率近似相同
1 | -3 # 模拟 NVE 系综 |
POMASS
元素的原子相对质量(数组)
默认缺省,会从 POTCAR 文件中读取
PMASS
为点阵自由度分配虚拟质量(单位 amu;NPT 系综的 Parinello-Rahman 算法)
默认值:1000
LANGEVIN_GAMMA、LANGEVIN_GAMMA_L
LANGEVIN_GAMMA (γ) 原子自由度的 Langevin 摩擦系数,LANGEVIN_GAMMA_L 点阵自由度的 Langevin 摩擦系数
NVT 系综,Langevin 热浴需设置 LANGEVIN_GAMMA;NPT 系综,Langevin 热浴需设置 LANGEVIN_GAMMA 和 LANGEVIN_GAMMA_L
对于所有的 non-Langevin 原子,γ 值为 0
LANGEVIN_GAMMA 默认是:0.0 x 元素种类数;LANGEVIN_GAMMA_L 默认值:0.0
LANGEVIN_GAMMA 建议设置值:10.0(与 POTIM 的乘积 = 10.0);LANGEVIN_GAMMA_L 建议设置值:1.0
KPAR、NPAR、NCORE
KPAR: 同时计算多少个 K 点(determines the number of k-points that are to be treated in parallel);默认值:1
NPAR:每个 K 点同时计算多少条能带/轨道(determines the number of bands that are treated in parallel);默认值:核数
NCORE: 一条带由多少个核来计算(determines the number of compute cores that work on an individual orbital);默认值:1;NCORE=总核数/KPAR/NPAR(NPAR 和 NCORE 只能设置其中之一)
VASP 计算里,平均而言,一个原子贡献 4 个价电子,4 个带
KPAR 最大可设为布里渊区不可约 K 点数,取决于 KPOINTS 文件设置及结构对称性
NPAR (或 NCORE) 和 KPAR 参数可以同时设置,对计算效率影响较大。并非所有可能的组合都能提供比默认值更好的计算效率
小体系并行:小体系的特点是原子数少,K 点较多,能带少;因此 KPAR 应尽可能设大, NPAR 可设为 1
中等体系并行:在中等规模的体系(20-100 个原子)中,情况会有所不同。此时,计算的不可约 k 点的数量很少(5-50),而电子能带的数量会很大(>100)
1 | # K 点、能带数目 示例 |
其他
- LDA + U:LDA+U - 计算材料学
1 | GGA = PE # 泛涵 |