ASE 使用
ASE 使用
介绍
atomic simulation environment (ASE):一系列用于设置、操作、运行、可视化及分析原子模拟的工具和 Python 模块
ASE 可支持的构型文件格式:File input and output — ASE documentation
ASE 通过
Calculators为不同的计算代码(DFT/MD)提供接口,Calculators与核心Atomsobject 和 ASE 中的许多可用算法一起使用;支持的Calculators如下:

ASE 版本 Release notes:查看版本更新细节
注意事项:
- ASE 网站中的源代码参数及注释与安装的 package 源码会有不一致的地方,以 pacakge 源码为准
- 无直接计算弹性常数的模块
- 很多变量的类型是
numpy.ndarray
参考资料
ase 教程(内容较详细):ASE tutorials;源码:GitHub - ASE-Workshop-2023/tutorial
ase tutorial:ASE_tutorial.ipynb
ase md 模拟:GitHub - PythonFZ/ase_md_example
弹性性质计算的 ASE 接口(弹性常数、EOS、声速;感觉一般):GitHub - jochym/Elastic: A module for ASE for elastic constants calculation.
基于 PAW 和 ASE 的 DFT code:GPAW: DFT and beyond within the projector-augmented wave method — GPAW
GitHub - AlexBoucherr/ASExVASP: A serie of script to perform calculations on VASP using the ASE
GitHub - jkitchin/dft-book: A book on modeling materials using VASP, ase and vasp
ase 结构 2D 和 3D 渲染:GitHub - chrisjsewell/ase-notebook: Highly configurable 2D (SVG) & 3D (threejs) visualisations for ASE/Pymatgen structures, within the Jupyter Notebook.
GitHub - superstar54/x3dase: X3D for Atomic Simulation Environment
ase symmetry 教程(内容一般):GitHub - ajjackson/ase-tutorial-symmetry: Tutorial notebook for symmetry features in ASE
lab5 有 ase NEB 计算:labutil/samples at master · bkoz37/labutil · GitHub
原子建模之ASE篇_哔哩哔哩_bilibili(主要生成纳米结构)
CompMatBook/Chapter04/4_2_辅助建模软件ASE生成纳米结构.ipynb at main · stanfordbshan/CompMatBook · GitHub
内容一般(能带计算 ASE Python 代码部分可参考): GitHub - WMD-group/ASE-Tutorials: Examples of using the Atomic Simulation Environment
安装
1 | pip install ase # 安装 |
使用
CLI
1 | # 开启 ase 补全(只适用于 bash,zsh 不行) |
其他
ase 缺陷计算 - 寻找最优的超胞形状:Tools for defect calculations — ASE documentation
interface 构建(较简单情况):Interface building - Manipulating atoms — ASE documentation
1 | # 振动分析 |
常用模块
ase.atoms
Atom 和 Atoms 是 ASE 的两个基本 Object, Atoms 由 Atom 构成
Atoms 本质上是 Atom 的 list,可以使用标序的方式来查看 Atom
需添加 calculator 才能使用的 methods:Adding a calculator
1 | from ase.atoms import Atoms |
- 晶体结构常用变量获取
1 | from ase.atoms import Atoms |
- 其他用法
1 | from ase.atoms import Atom, Atoms |
ase.build
- 结构建模
Bulk 晶体结构
1 | from ase.build import bulk |
Surface 表面模型
无法枚举出具有不同表面终端的所有表面(建议还是通过 Material Studio 或 pymatgen surface 模块或 atomsk 构建含该表面坐标的单胞,进而查看原子层数)
常见晶体结构(BCC、FCC、HCP、Diamond)的常见表面(100、110、111)构建函数,支持吸附位点(’ontop’, ‘bridge’, ‘hollow’ 等)
指定面指数切表面(比 pymatgen surface 模块好用)
注意事项:
- fcc100、fcc110、bcc100、hcp10m10、diamond100 总是返回正交胞
- fcc111、bcc110、bcc111、hcp0001 可返回非正交胞和正交胞两种(优先正交胞)
- fcc211 只返回正交胞;diamond111 只返回非正交胞
- size 设置有要求,一般是需要设置 y 方向的 size 数值为偶数
- root surface 是什么含义?
1 | from ase.build import ... |
- 其他
1 | from ase.build import add_vacuum, sort |
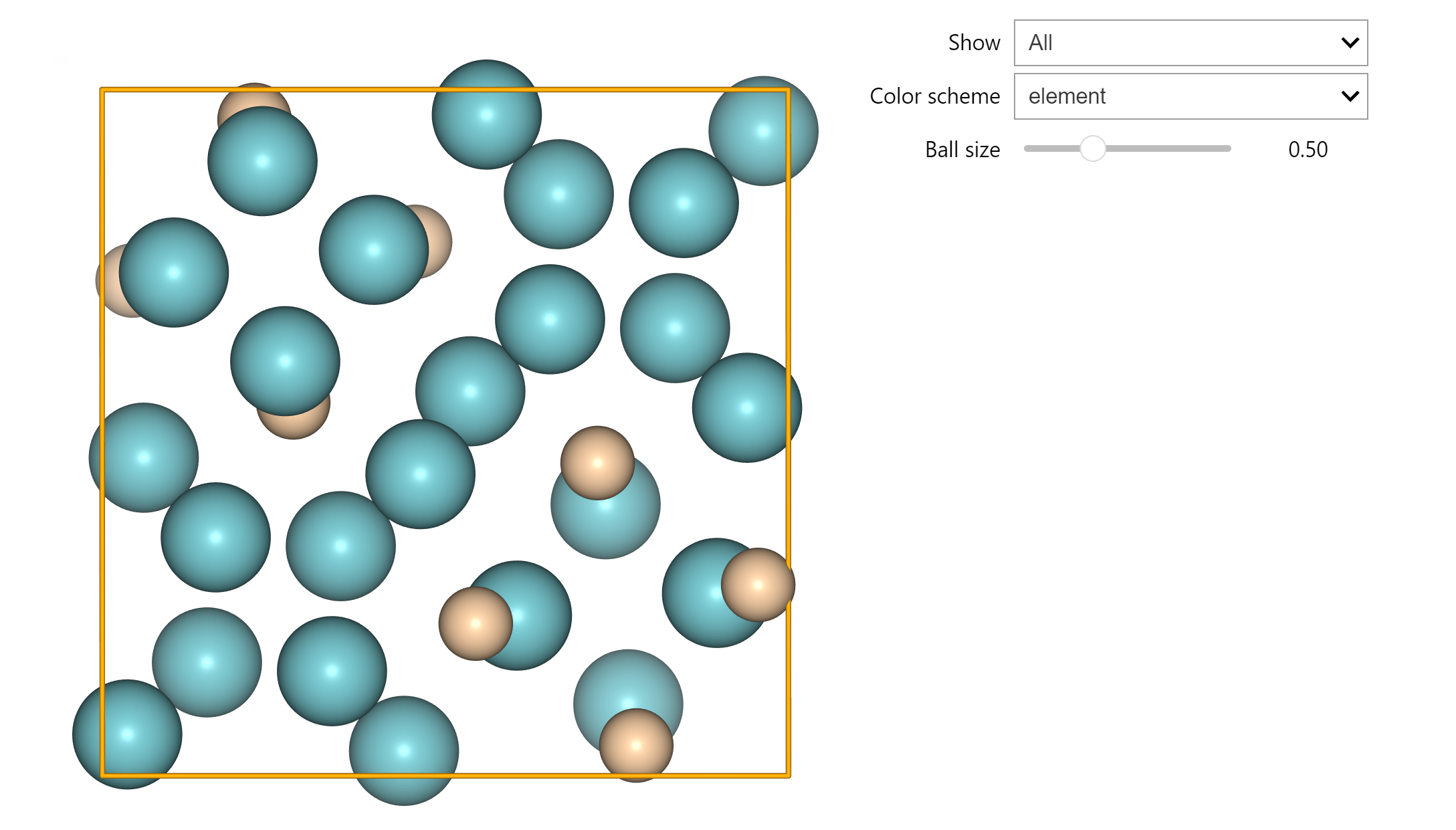
ase.visualize
- 构型可视化(建议在 Jupyter Notebook 中使用)
1 | # 方式 1 |
- nglview 组件效果图:
ase.cell
- 基矢/点阵
1 | from ase.cell import Cell |
ase.spacegroup
- 空间群
1 | from ase.spacegroup import Spacegroup, crystal, get_spacegroup |
ase.lattice
- 点阵
有生成 graphene 和 graphite 模块:ase/lattice/hexagonal.py · master · ase / ase · GitLab
Bravais lattices — ASE documentation
1 |
ase.io
构型文件读入、写出;可读入压缩格式文件内容;可支持的格式很多;File input and output — ASE documentation
部分构型文件格式只读 (R) 或只写 (W) 或可读入写入多帧构型 (RW+) 数据
1 | # 常用构型文件格式 读写情况 |
模块中的
read()函数可自动识别文件格式写法一:在
read()、write()函数中指定format参数,即具体构型文件格式
1 | from ase.io import read, write |
- 写法二:从
ase.io中导入具体构型文件格式的模块及其函数
1 | # LAMMPS data 格式 |
- 删除/重置 extxyz 文件中已有的 forces 和 info 信息:Issue with write_xyz (#276) · 议题 · ase / ase · GitLab
1 | atoms = ... |
ase.constraints
施加约束
注:pymatgen 只支持 ASE 中的 FixAtoms and FixCartesian,其他 constraints 在 Atoms 转 Structure 时不会被设置
1 | from ase.constraints import FixAtoms, FixedLine, FixedPlane |
ase.eos
- 执行 EOS 计算并拟合,得到体模量
1 | from ase.eos import calculate_eos |
- 根据 E-V 数据进行 EOS 拟合,获取平衡能量,体积和体模量;并绘制 EOS 拟合曲线
1 | from ase.eos import EquationOfState |
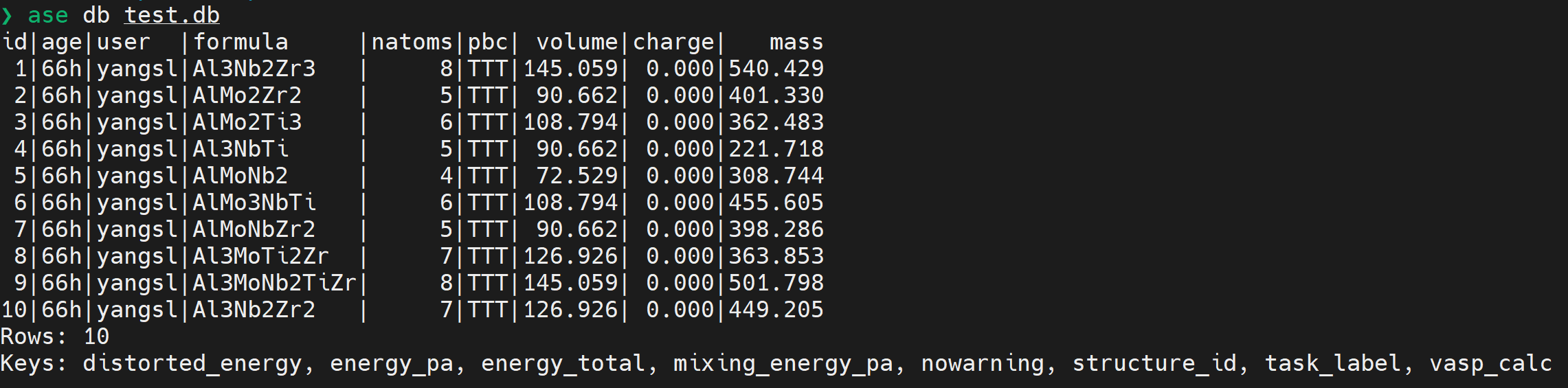
ase.db
ASE databases 文件
db 文件 TUI 接口:GitHub - steenlysgaard/texase: A textual user interface for ASE databases
db.select(sort=...)中的sort为含 key 的 str,含-时,降序
1 | from ase.db import connect |
ase.optimize
- 优化器(优化算法)
1 | from ase.optimize import QuasiNewton # 等于 BFGSLineSearch |
ase.data
- 元素周期表中的元素相关数据
1 | from ase.data import ... |
ase.units
ASE 中的物理单位,电子伏特 eV、埃 Å,开尔文 K 和原子质量单位定义为 1.0
1 | from ase.units import Bohr, Hartree, eV, kJ, mol, GPa, _Nav |
ase.calculators
ASE 支持的 calculators:Supported Calculators — ASE documentation
ASE 原生 Python 实现的 caculators:EMT (effective medium theory),EAM,Lennard-Jones,Morse 和 HarmonicCalculator
VASP
设置 VASP 执行命令和赝势路径(在
~/.{bash,zsh}rc或在 Python 脚本中设置环境变量)
1 | export ASE_VASP_COMMAND="mpirun path/vasp_std" |
1 | import os |
- ASE VASP Calculator 赝势不同泛涵目录命名
1 | potpaw_PBE # PBE |
- 示例代码
1 | from ase.calculators.vasp import Vasp |
1 | from ase.calculators.vasp import Vasp |
LAMMPS
- 示例代码
1 | from ase.calculators.lammpsrun import LAMMPS |
GPAW
- 示例代码
1 | from gpaw import GPAW |
设置单点能
1 | # 设置单点能 |
1 | # 使用多种 calculator |
ase.phasediagram
相图绘制(2 维,3 维):Phase diagrams and Pourbaix diagrams — ASE documentation
没有 pymatgen 对应的模块功能丰富且绘图效果好看
ase.cluster
- 纳米颗粒/团簇
ase.geometry
- rdf 计算
1 | from ase.geometry import ... |
ase.neb
NEB 计算、拟合、分析与绘制
1 | from ase.mep import NEB, NEBTools |
ase.md
ASE 中的 MD 相关算法
ASE 中的两种常见控压(压浴)方法:Parrinello-Rahman、Berendsen
使用 ASE md 模块执行 MD 示例:
1 | from ase.md import MDLogger |
ase.phonons
- 有限位移法计算声子谱
1 | # 示例 |
ase.filters
1 | # 结构优化,用于非 VASP 其他 Calculator(如 GPAW) |
ase.neighborlist
- 近邻列表
1 | from ase.neighborlist import NeighborList |